

Определение, этиология, классификация, диагностика, лечение
Нервно-мышечные заболевания представляют собой группу заболеваний мышц скелета и расположенных в конечностях нервов (моторных, сенсорных), во время которых первичный патологический процесс отмечается непосредственно в мышце или нерве. При этом центральная нервная система не повреждена. Нервно-мышечные заболевания, занимающие значительное место в клинической неврологии, также известные как заболевания периферийной нервной системы.
Классификация нервно-мышечных заболеваний происходит согласно анатомическому распространению патологического процесса. Патологические изменения отмечаются:
- В поперечнополосатой мышце (мышечные дистрофии, врожденные миопатии, метаболические и воспалительные миопатии);
- В моторной клетке, расположенной в спинном мозге (атрофия спинных мышц);
- В нерве (наследственные полиневропатии, воспалительные невропатии);
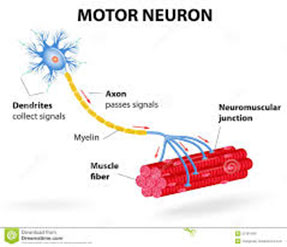
- В синапсе – в месте передачи импульса из нерва в мышцу (миастения, синдром врожденной миастении, неонатальная миастения).
1. Мышечные дистрофии – самой распространенной формой являются мышечная дистрофия Дюшенна и Беккера, которые вызваны отсутствием или значительным снижением в мышцах белка дистрофина. В таком случае патологический процесс включает мышечное волокно, в то время как расположенные в спинном мозге нервные моторные клетки, выходящие из спинного мозга нервы и нервные окончания не повреждены. Болезнь встречается среди мальчиков и передается генетически. Болезнь Дюшенна начинается до 5 лет. Первым признаком является усложненность подъема по лестницам и вставания с пола.
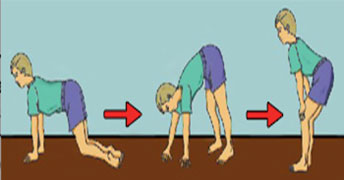
Походка становится утиной. Характерно увеличение мышц голени. В голенных сосудах развиваются контрактуры. В возрасте 8-12 лет пациент теряет способность передвигаться самостоятельно. Лечение кортикостероидами продлевает способность самостоятельного передвижения на 3-4 года. У большого числа больных отмечаются умственные проблемы, кардиомиопатия.
Мышечная дистрофия Беккера начинается в более позднем возрасте, после 8-10 лет. Клинические признаки те же, что и при дистрофии Дюшенна. Во время этой формы пациент дольше сохраняет способность самостоятельного передвижения.
Диагностика: определение в крови фермента креатинкиназа – значительно увеличен при обеих формах.
Электронейрограммное обследование – миопатические записи.
Генетическое исследование. Биопсия мышцы.
Лечение в основном ориентируется на сохранение функциональной независимости и качества жизни. Физиотерапия направлена против развития контрактур и сколиоза. Использование ортозов и вовремя проведенное хирургическое вмешательство на сухожилия улучшает хождение. В случае необходимости проводится электрокардиографический и эхокардиографический мониторинг. Для предотвращения дыхательной недостаточности, вызванной слабостью дыхательных мышц, пациенту необходимо проводить спирометрию – оценку вдоха-выдоха. В случае необходимости нужна дополнительная вентиляция.
Сопутствующие миопатии – выявление происходит в младенчестве или раннем детстве по общей вялости мышц. Характерны гистохимические и ультраструктурные изменения мышц, медленное прогрессирование. Важен такой клинический признак, каким являются опущенные веки, кардиомиопатия. Существует классификация сопутствующих миопатий, основанная на генетических и морфологических изменениях, согласно которой выделены следующие миопатии:
- С накоплением белка;
- Миопатии с осью;
- Миопатии с центральным ядром;
- Миопатии с изменением размера волокон;
- Диагностика – креатинкиназа в большинстве случаев в норме;
- Элеткроневромиограммное обследование в норме.
Установление диагноза происходит путем биопсии мышцы.
Лечение в основном ориентируется на реабилитацию, основной целью которой является сохранение функциональной независимости и качества жизни. Важна постоянная оценка дыхательной системы, сколиоза и способности самостоятельного передвижения.
2. Спинальная мышечная атрофия представляет собой наследственное прогрессирующее заболевание расположенных в спинном мозге двигательных клеток (мотонейронов), которая протекает с мышечной слабостью и вызвана медленным вырождением (дегенерацией) этих клеток.
Выделяется 3 основных типа этой болезни:
- Болезнь Вердниг-Хоффмана – выявление осуществляется в основном в первые месяцы жизни. Ребенку трудно держать голову, слабые плач и кашель. Характерны частые опоздания во время кормления. Выражена слабость дыхательных мышц. К 7-8 месяцам не сидит самостоятельно.
- Промежуточная форма детства – слабость развивается сравнительно поздно. Возможно самостоятельное сидение, развиваются сосудистые контрактуры, отмечается опоздание пищи.
- Болезнь Кугельбер-Виландера – характеризуется поздним началом. Пациенты могут самостоятельно ходить. Наблюдается сколиоз, сложность при глотании и кашле. Характерна симметричная слабость проксимальных мышц. Течение болезни прогрессирующее, темп снижается с возрастом.
Диагностика основывается на:
- Электроневромиограммном обследовании – неврогенные записи;
- Определении креатинкиназы в крови;
- Генетическом обследовании – тест делеции.
Уход и лечение пациентов с болезнью Вердниг-Хоффмана включает трахеостому, вспомогательную вентиляцию и кормление через зонд. Для других форм лечение симптоматично и направлено на предотвращение деформации позвоночника и нижних конечностей.
Важно своевременное определение и правильное управление указанных болезней.
3. Периферийные невропатии: патологические изменения могут повреждать нервные волокна (моторные и сенсорные) в конечностях. В зависимости от того, сколько нервов и какая анатомическая часть нерва повреждены,
Периферийные невропатии делятся на следующие виды: полиневропатия; мононевропатия; множественный мононеврит; радикулопатия; полирадикулопатия; плексопатия (бок, поясница).
Самым частым дегенеративным заболеванием периферийной нервной системы является периферийная моторная и сенсорная полиневропатия, которая известна как болезнь Чаркот-Мари-Тут. В расположенных в конечностях нервах развиваются дегенеративные изменения, что вызывает затрудненность ходьбы, частые падения, атрофию мышц, а позднее деформацию стопы, что выборочно характерно для этой болезни.
Классификация опирается на тип распространения болезни и вид наследственной передачи. Развитие молекулярной генетики позволило выделение нескольких типов наследственной моторной и сенсорной невропатии.
Диагноз основывается на электроневромиограммном исследовании (характерно падение скорости электрического импульса). В неопределенных случаях прибегают к молекулярно-генетическому обследованию.
Лечение основывается на командном наблюдении различных специалистов (физиотерапевт, оккупационный терапевт, ортопед, невролог), реабилитации, превенции контрактур. В случае фиксированных контрактур прибегают к хирургическому лечению.
4. Миастения – самое частое заболевание нервно-мышечного синапса в детском возрасте, является автоиммунным. Его причинами являются уменьшение рецепторов принятия импульса в синапсе – месте передачи импульса из нерва в мышцу. Уменьшение рецепторов обуславливается выработанными в организме антителами, этиология которых неизвестна. Симптомы проявляются в возрасте старше одного года. У пациентов наблюдается слабость, быстрая утомляемость, особенно во второй половине дня, затрудненное жевание и глотание, опущенные веки, раздвоенное зрение. В случае обобщенных форм миастении слабость распространяется также на мышцы конечностей. Установлена связь миастении с другими аутоиммунными процессами – патологией щитовидной железы и заболеванием вилочковой железы (тимомы). Тяжесть болезни выявляется на сравнительно раннем этапе. Существует отдельная классификация клинических форм миастении (классификация Осермана).
Диагностика основывается на выявлении антител в крови относительно рецепторов ацетилхолина и электроневромиограммном обследовании (наличие декремента при повторной стимуляции).
При лечении в основном используются медикаменты, которые содержат ацетилхолинестеразу (неостигмин, пиридостигмин), а также плазмаферез и внутривенный иммуноглобулин, иммуносупрессоры. В некоторых случаях прибегают к удалению вилочковой железы.